
Anisotropic displacement parameters from dispersion-corrected DFT methods and their experimental validation by temperature-dependent X-ray diffraction†
Abstract
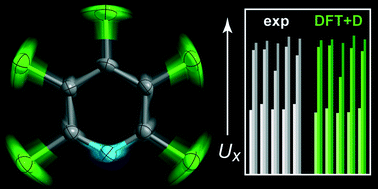
In chemical crystallography, the thermal motion of scattering centres is commonly described by anisotropic displacement parameters (ADPs). Very recently, it has been shown that ADPs are not only accessible by diffraction experiments but also via theory: this emerging approach seems promising but must be thoroughly tested. In this study, we have performed specifically tailored X-ray diffraction (XRD) experiments in fine steps between 100 and 300 K which allow detailed comparison to ab initio data from dispersion-corrected density functional theory (DFT) combined with periodic lattice-dynamics. The compound chosen for this study, crystalline pentachloropyridine (C5NCl5), is well suited for this purpose: it represents a molecular crystal without H atoms, thus posing no challenge to XRD; its solid-state structure is controlled by dispersion and halogen-bonding interactions; and the ADPs associated with the peripheral Cl atoms show strong temperature dependence. Quality criteria in direct and in reciprocal space prove that ADPs are predicted with high confidence for the temperature range between 100 and 200 K, and that several economic dispersion corrections to DFT can be reliably employed for this purpose. Within the limits we have explored here, the ab initio prediction of ADPs appears to be a facile and complementary tool, especially in those cases where diffraction data cannot provide a straightforward model for thermal motion.
 Please wait while we load your content...
Please wait while we load your content...

