
A polar/π model of interactions explains face-to-face stacked quinoid rings: a case study of the crystal of potassium hydrogen chloranilate dihydrate†
Abstract
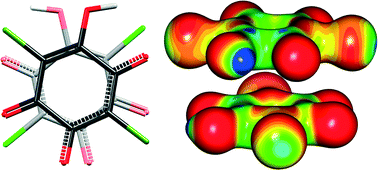
The nature of interactions between face-to-face staggered stacked quinoid rings with π-systems, observed with a short inter-ring centroid⋯centroid distance, is analyzed by experimental and theoretical methods. Charge density studies based on X-ray diffraction and DFT calculations, complemented by impedance spectroscopy, were employed to define the electronic and structural characteristics of the quinoid rings responsible for their interactions within the crystal packing. The crystal packing is mainly stabilized by strong electrostatic interactions between the K+ cation and the hydrogen chloranilate anion. The proximity and orientation of the stacked quinoid rings in parallel glide-plane arrangement are partly governed by the non-covalent interactions within the dimer. The estimated contribution of dispersion energy to the stacking of the rings about −10 kcal mol−1, as calculated by DFT methods, is comparable to medium–strong hydrogen bonding. The electronic structure of 3,6-dichloro-2,5-dihydroxyquinone monoanion exhibits alternating electron-rich and electron-poor regions. The calculated electrostatic energy shows variations with the relative orientation of rings within dimers reaching ca. 10–15 kcal mol−1. Thus, the nature of interactions between π-systems of quinoid rings can be described by a polar/π model where electrostatic complementarity plays a determinant role in π-stacking orientation. These interactions have great potential in crystal engineering and may be employed in the design of functional materials.
 Please wait while we load your content...
Please wait while we load your content...

