
Using dispersion-corrected density functional theory to understand supramolecular binding thermodynamics†
Abstract
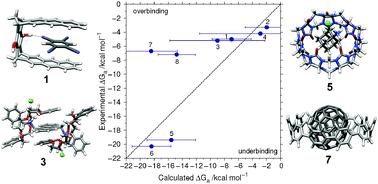
A recently published theoretical approach employing a nondynamic structure model using dispersion-corrected density functional theory (DFT-D3) to calculate equilibrium free energies of association (Chem. Eur. J., 2012, 18, 9955–9964) is illustrated by its application to eight new supramolecular complexes. We compare with experimentally known binding constants which span the range from −3.3 to −20.3 kcal mol−1. The mean deviation of calculated from measured ΔGa results in 0.4 kcal mol−1, the mean absolute deviation in 1.8 kcal mol−1 excluding two outliers for which the computed solvation free energies are identified as the largest error source. A survey of previous applications of the theoretical approach and related computational studies is given underlining its good accuracy. It is concluded that structures and gas phase interaction energies can be computed routinely with good to high accuracy (relative errors for interaction energies of 5–10%) for complexes with about 200–300 atoms.
 Please wait while we load your content...
Please wait while we load your content...

