
Bimetallic catalysis in the highly enantioselective ring–opening reactions of aziridines†
Abstract
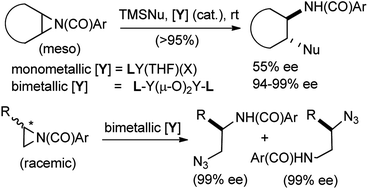
Bimetallic yttrium- and lanthanide-salen complexes, readily prepared from commercially available metal isopropoxides, 2-dimethylaminoethanol, 1,1′-binaphthyl-2,2′-diamine and 2-hydroxy-3-methoxybenzaldehyde (3 steps) catalyze highly enantioselective ring opening (∼90–99% ee) reactions of meso-N-4-nitrobenzoyl aziridines by TMSCN and TMSN3. The TMSN3-mediated reactions give the highest enantioselectivities reported to date for several prototypical aziridines. Selectivity in a related ring opening by silyl isothiocyanates depends on the substituents on silicon, larger tBuPh2SiNCS giving the best selectivities, especially when an yttrium or ytterbium complex is used as the catalyst. The bimetallic yttrium complex also effects unprecedented regiodivergent parallel kinetic resolution of racemic monosubstitued aziridines upon reaction with TMSN3. In these reactions, each of the enantiomers undergoes nucleophilic addition of azide at different carbons giving two products in nearly enantiomerically pure form. To explain the dramatic differences in the selectivities between the mono- and bimetallic catalysts in these reactions, a mechanism that involves activation of both the electrophile (aziridine) and the nucleophile (azide or cyanide) at two different metals of the bimetallic complex is proposed. Molecular weight determination by vapor pressure osmometry, diffusion ordered NMR spectroscopy (DOSY) and kinetic studies, which suggest first order dependence on the concentration of the catalyst, provide strong support for this proposal.
 Please wait while we load your content...
Please wait while we load your content...

