
Interactions of the aquated forms of ruthenium(iii) anticancer drugs with protein: a detailed molecular docking and QM/MM investigation
Abstract
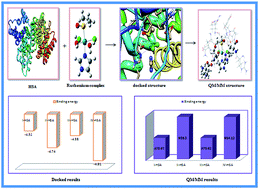
Interaction of monoaqua and diaqua ruthenium complexes such as [trans-RuCl3(H2O)(3H-imidazole)(dmso-S)] I, [trans-RuCl2(H2O)2(3H-imidazole)(dmso-S)]+1 II, [trans-RuCl3(H2O)(4-amino-1,2,4-triazole)(dmso-S)] III and trans-RuCl2(H2O)2(4-amino-1,2,4-triazole)(dmso-S)]+1 IV, which are formed after intracellular aquation of their respective complexes, with human serum albumin (HSA) has been computationally investigated by molecular docking and two layer QM/MM hybrid methods. The computed binding energy of monoaqua adduct I–HSA and III–HSA evaluated by docking simulation are found to be −4.52 kcal mol−1 and −4.58 kcal mol−1 whereas the binding energy of diaqua adducts II–HSA and IV–HSA are evaluated to be −4.74 kcal mol−1 and −4.91 kcal mol−1, respectively. Docking results also show that the ruthenium atoms of all the complexes are actively involved in coordination with histidyl nitrogen atoms in the active site of protein. In addition, in order to probe the stabilities of monoaqua and diaqua ruthenium complexes in the active site of protein, we have calculated their energetic by two layer QM/MM method. QM/MM study suggests higher stability of diaqua adduct, II–HSA. The stability of adducts varies in the order: II–HSA > IV–HSA > I–HSA > III–HSA. Binding energy values of all the complexes increase with the incorporation of solvent effect. Thus molecular docking and QM/MM results show that ruthenium complexes interact with the protein receptor more rapidly after their second hydrolysis. Hence, docking as well as ONIOM results will be highly beneficial for providing insight into the molecular mechanism of ruthenium complexes with protein receptor.
 Please wait while we load your content...
Please wait while we load your content...

