
Theoretical investigation on unimolecular decomposition of malonic acid: a potential sink for ketene†
Abstract
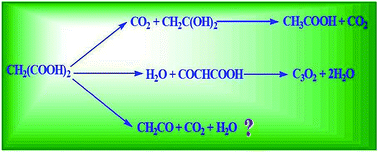
DFT and ab initio calculations are performed to study the unimolecular decmposition pathways of malonic acid. The reaction is explored along three plausible decomposition pathways. The calculated results shows that decarboxylation is a two step process with an energy barrier of 32.16 kcal mol−1 at the CCSD(T)/6-311++G(d,p) level which is in reasonable agreement with available data. In contrast to the usual interpretations, novel features of this study are dehydration and in situ decarboxylation–dehydration pathways that involve energy barriers of 67.13 and 44.75 kcal mol−1, respectively at the CCSD(T)/6-311++G(d,p) level leading to the formation of carbon-suboxide and ketene along with conventional H2O and CO2. The thermal rate constants for the above decomposition pathways are also evaluated using Canonical Transition State Theory at 298 K.
 Please wait while we load your content...
Please wait while we load your content...

