
Spectroscopic study and electronic structure of prototypical iron porphyrins and their μ-oxo-dimer derivatives with different functional configurations†
Abstract
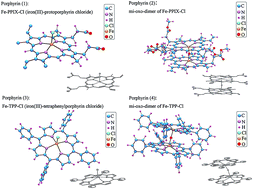
Metalloporphyrins are networking molecules with strong internal N–H hydrogen bonds that may be used to substitute a metal ion inside a porphyrin ring, forming a metallo complex. The understanding of electronic structures and charge dynamics of porphyrin based molecular architectures is mandatory to clarify biological functions, catalytic processes, and opto-electrical responses in which these molecules are involved. We present here a systematic analysis of the electronic structures and the charge dynamics of two different iron-porphyrins (i.e. protoporphyrin IX and meso-tetraphenylporphine) with different functional architectures. We investigated these prototypical porphyrins and their μ-oxo-dimer derivatives by means of Fe K-edge X-ray Absorption Near-Edge Spectroscopy (XANES) combined with theoretical calculations. The electronic structure, namely the partial projected density of states and the polarization components were discussed in terms of orbital hybridizations among metal and local ligands. Data show that hydrogens are electron donors while the central metal irons accept electrons. Moreover, the metal axial ligands exhibit different electron behaviors: donors for Cl in prototypical porphyrins and acceptors for O in μ-oxo-dimer derivatives. Actually, the charge dynamics are affected by local metal ligands, but also strongly depend on the mid-range atomic ordering of the porphyrins network. The charge dynamics, evaluated from the self-consistent local potential, is associated with charge transfer mechanisms involving interactions with the axial ligands as well as with the substituents. The quantum chemical topology analysis of the electron localization function (ELF) has been used to identify the distribution of the electron pairs. They are localized around Cl atoms regardless of porphyrin configurations. Charge dynamics and electron localization are fundamental information for a deep understanding of the role of porphyrin and porphyrin-like molecules in a wide range of molecular biophysical mechanisms and in materials science processes.
 Please wait while we load your content...
Please wait while we load your content...

