
Metal ionic size directed complexation in manganese(ii) coordination chemistry: efficient candidates showing phenoxazinone synthase mimicking activity†
Abstract
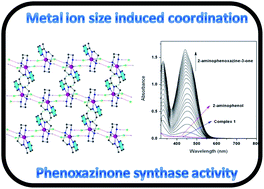
The present report describes the syntheses and structural characterizations of two new mononuclear manganese(II) complexes, [Mn(L1)Cl2]·2MeOH (1) and [Mn(L2)Cl2] (2), in which L1 and L2 are tetradentate ligands. Although the previous studies described that ligand L1 exclusively binds the metal centers (Fe2+, Ni2+ and Zn2+) in the acyclic isomeric form (Schiff dibasic) of the ligand, in the present investigation it selectively binds Mn2+ ion in its cyclic (hexahydropyrimidine) analogue during the complexation reaction as evidenced by X-ray crystallography. The structure of 2 is quite interesting as it shows that one arm of the Schiff dibasic form of the ligand has been hydrolyzed, suggesting that these types of ligands with the Schiff dibasic form are incapable of yielding the stable manganese(II) complexes. The metal ionic size directed hydrolysis of one arm of the ligand has been confirmed by IR spectral studies. Both the complexes are reactive towards the oxidation of o-aminophenol (OAPH), and their relative catalytic efficiencies can be clearly explained by considering the steric contribution from the ligands and the electrochemical responses of the metal center. From the experimental data, a nice correlation, wherein the lower the E1/2 value the higher the catalytic activity, can be drawn between E1/2 and Vmax of the complexes. The kinetics study exhibited a deuterium kinetic isotope effect in the catalytic oxidative coupling of two moles of OAPH by O2 as evidenced by the 1.6 times rate retardation in the deuterated solvent, suggesting hydrogen atom transfer in the rate-determining step from the substrate hydroxy group to the metal-bound superoxo species.
 Please wait while we load your content...
Please wait while we load your content...

