
First-principles investigation on dimerization of metal-encapsulated gold nanoclusters
Abstract
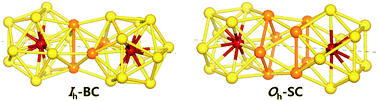
Density functional theory is used to study dimerization of metal-encapsulated gold nanoclusters M@Au12 (M = W, Mo) with Ih or Oh symmetry, and their structural and electronic properties. To determine the most stable dimer structure in each case, various configurations are considered. We find that during dimerization, gold atoms near the interface tend to form inter-cluster triangular bonds, which stabilize two monomer clusters by about 3.3–3.5 eV. The dimerization along a specific axis selected as the z axis causes the symmetry reduction of each M@Au12 cluster resulting in the modification of electronic structures. It is found that all the stable dimers exhibit a much smaller HOMO–LUMO gap than those of their comprising monomers. Such a gap decrease is mainly attributed to the dz2 orbital splitting of the central atoms owing to dimerization. We also calculate the vibrational modes and the corresponding IR-active spectra, which are distinguishable for different dimer configurations. In addition, we find that the IR-active modes of the Oh-based dimer structures appear to be red-shifted in comparison to those of Ih-based ones. Thus, the IR spectra may be utilized experimentally to discriminate dimer configurations with different central metal atoms and/or dissimilar structural symmetries.
 Please wait while we load your content...
Please wait while we load your content...

