
Revised electronic structure, Raman and IR studies of AB2H2 and ABCH (A = Sr, Ba; B = Al, Ga; C = Si, Ge) phases†
Abstract
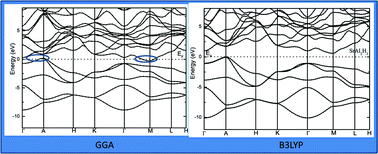
Modern density functional theory (DFT) provides an extremely valuable tool for predicting structures and energetics of new materials for both finite and periodic systems. In the present work the electronic structure, chemical bonding, and spectroscopic properties of AB2H2, and ABCH (A = Sr, Ba; B = Al, Ga; C = Si, Ge) phases have been studied by state-of-the-art density-functional calculations. In contrast with the previous theoretical studies, present electronic structures calculation using hybrid B3LYP reveal that AB2H2, phases are indirect bandgap semiconductors with estimated bandgap vary from 0.324 to 0.495 eV. In general, B3LYP gives a reasonable description of the electronic structure of semiconductors, semi-ionic oxides, sulfides, transition metal oxides and the ionic oxide. Similar to the AB2H2 phases ABCH phases are also indirect bandgap semiconductors and the magnitude is much higher than for the AB2H2 phases. We have also simulated the Raman and IR spectra, and calculated the NMR related parameters such as isotropic chemical shielding, quadrupolar coupling constant, and quadrupolar asymmetry for the AB2H2, and ABCH phases.
 Please wait while we load your content...
Please wait while we load your content...

