
Stereocontrolled lithiation/trapping of chiral 2-alkylideneaziridines: investigation into the role of the aziridine nitrogen stereodynamics†‡
Abstract
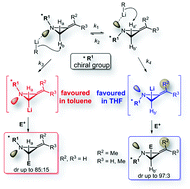
The origin of the stereoselectivity in the lithiation/trapping of 2-alkylideneaziridines bearing a chiral group as the nitrogen substituent was investigated. Optimal reaction conditions were discovered by in situ FT-IR monitoring. In addition, it has been found that the solvent and the alkene substitution pattern are important factors able to impart a switch in stereoselectivity. While lithiation of the alkylideneaziridine ring flanked by either a fully substituted or a Z-configured alkene pendant occurs stereoselectively in THF, in contrast unsubstituted 2-methyleneaziridine undergoes lithiation in toluene with the opposite sense of stereoinduction. Lithiation experiments, on deuterium labelled 2-alkylideneaziridines, confirmed the configurational stability of the lithiated intermediates. A model based on complexation and proximity effects was proposed to rationalize the reactivity. This model assumes that slowly equilibrating N-invertomers undergo deprotonation (lithiation) at different rates and that the stereochemical outcome is established during the deprotonation step.
 Please wait while we load your content...
Please wait while we load your content...

