
Quantum mechanistic insights on aryl propargyl ether Claisen rearrangement†
Abstract
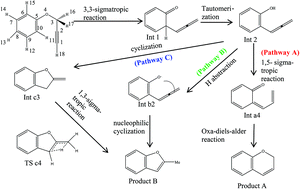
The mechanism of aryl propargyl ether Claisen rearrangement in gas and solvent phase was investigated using DFT methods. Solvent phase calculations are carried out using N,N-diethylaniline as a solvent in the PCM model. The most favorable pathways involve a [3,3]-sigmatropic reaction followed by proton transfer in the first two steps and then deprotonation or [1,5]-sigmatropic reaction. Finally, cyclization yields benzopyran or benzofuran derivatives. The [3,3]-sigmatropic reaction is the rate-determining step for benzopyran and benzofuran with ΔG‡ value of 38.4 and 37.9 kcal mol−1 at M06/6-31+G**//B3LYP/6-31+G* level in gas and solvent phase, respectively. The computed results are in good agreement with the experimental results. Moreover, it is found that the derivatives of aryl propargyl ether proceeded Claisen rearrangement and the rate-determining step may be shifted from the [3,3]-sigmatropic reaction to the tautomerization step. The NBO analysis revealed that substitution of the methyl groups on the aliphatic segment has decreased the stabilization energy E(2) and favors the aryl propargyl ether Claisen rearrangement.
 Please wait while we load your content...
Please wait while we load your content...

