
Comparative analysis of cross strand aromatic–Phe interactions in designed peptide β-hairpins†
Abstract
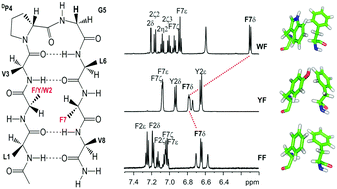
The mode(s), geometry and strength of interaction of the three aromatic amino acids, namely Phe, Tyr and Trp, with the benzyl side chain of Phe, at the non-hydrogen bonding position of designed model octapeptide β-hairpins, nucleated by the central DPro–Gly turn, have been examined. In the absence of solvent-driven hydrophobic forces, the extent of contribution of such interactions indicates that the stereospecific face-to-edge (FtE) geometry of aromatic rings is most stabilizing in the Trp–Phe pair. In contrast, our study shows that the Tyr–Phe pair exhibits the weakest interaction energy, despite its abundance in protein structures. The contribution of aromatic interactions as opposed to the influence of spatial proximity to electron-rich groups, to the observed anomalous backbone and side chain chemical shifts, has also been delineated. Our findings indicate that the Trp–Phe pair contributes an additional ∼0.9 kcal mol−1 and ∼1.3 kcal mol−1 towards scaffold stabilization, when compared with the Phe–Phe and Tyr–Phe pair, respectively, even in an amphipathic solvent such as methanol. Detailed NMR analysis of backbone resonances, as well as the extent of pronounced anomalous chemical shifts, indicates that the strength of aromatic interactions with Phe follows the order Trp > Phe > Tyr. Furthermore, the advantages of Trp–Phe or Phe–Phe pairs as alternative structure stabilizing elements are also highlighted.
 Please wait while we load your content...
Please wait while we load your content...

