
A multiscale approach to determine binding energy distribution on a strained surface
Abstract
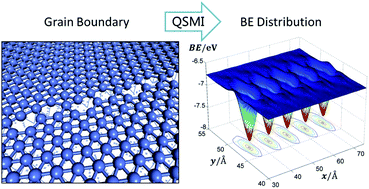
A multiscale approach was developed by combining ab initio calculations with classical molecular mechanics (MM) simulations to investigate the adsorption and diffusion of an adatom on a strained and/or defective surface. Specifically, the binding energy of the adatom was calculated as a function of the local substrate strain near the adsorption site by an ab initio method and the strain distribution of a large defective surface was calculated by the MM method. Then a map of the binding energy of the adatom on a large defective surface was derived by bridging the DFT calculated binding energy and the MM determined strain distribution. As an example, the approach is applied to explore the adsorption and diffusion of a carbon atom on the Ni(111) surfaces with dislocations and grain boundaries, respectively. This approach bridges models of different length scales and can be extended to systems with an uneven distribution of strain or curvature.
 Please wait while we load your content...
Please wait while we load your content...

