
Mechanistic insights into B–H bond activation with the high-valent oxo-molybdenum complex MoO2Cl2†
Abstract
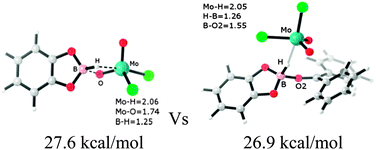
The B–H bond activation and the catalytic hydroboration of carbonyl compounds by the high-valent oxo-molybdenum complex MoO2Cl2 were theoretically investigated to determine the underlying mechanism. Our calculation results indicate an unique path – the ionic mechanistic pathway involving the heterolytic cleavage of the B–H bond competes with the [2+2] addition pathway, which involves the addition of the B–H bond across one of the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. The rate-determining free energy barriers for the ionic mechanistic pathway are calculated to be 26.9 kcal mol−1, 25.0 kcal mol−1 and 23.7 kcal mol−1 for diphenylketone, benzaldehyde and acetophenone, respectively. These values are energetically slightly favorable than the [2+2] addition mechanism by ∼1–3 kcal mol−1. Furthermore, it is worth noting that the carbonyl compounds bearing the electron donation group will induce a better activity toward the ionic mechanistic pathway.
O bonds. The rate-determining free energy barriers for the ionic mechanistic pathway are calculated to be 26.9 kcal mol−1, 25.0 kcal mol−1 and 23.7 kcal mol−1 for diphenylketone, benzaldehyde and acetophenone, respectively. These values are energetically slightly favorable than the [2+2] addition mechanism by ∼1–3 kcal mol−1. Furthermore, it is worth noting that the carbonyl compounds bearing the electron donation group will induce a better activity toward the ionic mechanistic pathway.
 Please wait while we load your content...
Please wait while we load your content...

