
Altered transition metal homeostasis in Niemann–Pick disease, type C1†
Abstract
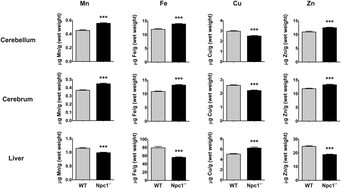
The loss of NPC1 protein function is the predominant cause of Niemann–Pick type C1 disease (NP-C1), a systemic and neurodegenerative disorder characterized by late-endosomal/lysosomal accumulation of cholesterol and other lipids. Limited evidence from post-mortem human tissues, an Npc1−/− mouse model, and cell culture studies also suggest failure of metal homeostasis in NP-C1. To investigate these findings, we performed a comprehensive transition metal analysis of cerebrospinal fluid (CSF), plasma and tissue samples from human NP-C1 patients and an Npc1−/− mouse model. NPC1 deficiency in the Npc1−/− mouse model resulted in a perturbation of transition metal homeostasis in the plasma and key organs (brain, liver, spleen, heart, lungs, and kidneys). Analysis of human patient CSF, plasma and post-mortem brain tissues also indicated disrupted metal homeostasis. There was a disparity in the direction of metal changes between the human and the Npc1−/− mouse samples, which may reflect species-specific metal metabolism. Nevertheless, common to both species is brain zinc accumulation. Furthermore, treatment with the glucosylceramide synthase inhibitor miglustat, the only drug shown in a controlled clinical trial to have some efficacy for NP-C1, did not correct the alterations in CSF and plasma transition metal and ceruloplasmin (CP) metabolism in NP-C1 patients. These findings highlight the importance of NPC1 function in metal homeostasis, and indicate that metal-targeting therapy may be of value as a treatment for NP-C.
 Please wait while we load your content...
Please wait while we load your content...