
Encompassing receptor flexibility in virtual screening using ensemble docking-based hybrid QSAR: discovery of novel phytochemicals for BACE1 inhibition†
Abstract
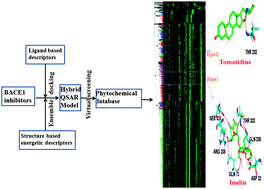
Mimicking receptor flexibility during receptor–ligand binding is a challenging task in computational drug design since it is associated with a large increase in the conformational search space. In the present study, we have devised an in silico design strategy incorporating receptor flexibility in virtual screening to identify potential lead compounds as inhibitors for flexible proteins. We have considered BACE1 (β-secretase), a key target protease from a therapeutic perspective for Alzheimer's disease, as the highly flexible receptor. The protein undergoes significant conformational transitions from open to closed form upon ligand binding, which makes it a difficult target for inhibitor design. We have designed a hybrid structure–activity model containing both ligand based descriptors and energetic descriptors obtained from molecular docking based on a dataset of structurally diverse BACE1 inhibitors. An ensemble of receptor conformations have been used in the docking study, further improving the prediction ability of the model. The designed model that shows significant prediction ability judged by several statistical parameters has been used to screen an in house developed 3-D structural library of 731 phytochemicals. 24 highly potent, novel BACE1 inhibitors with predicted activity (Ki) ≤ 50 nM have been identified. Detailed analysis reveals pharmacophoric features of these novel inhibitors required to inhibit BACE1.
 Please wait while we load your content...
Please wait while we load your content...