
Charting the mechanism and reactivity of zirconium oxalate with hydroxamate ligands using density functional theory: implications in new chelate design
Abstract
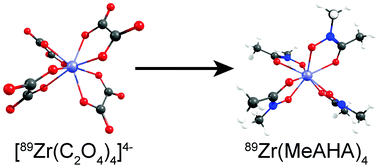
The reaction of [89Zr(C2O4)4]4− with the tris-hydroxamate ligand desferrioxamine B (DFO) provides the basis of radiolabelling biological vectors such as antibodies and proteins with the radionuclide 89Zr for positron emission tomography imaging. In this work, density functional theory methods were used to investigate the mechanism of reaction from [Zr(C2O4)4]4− to Zr(MeAHA)4 by ligand substitution with N-methyl acetohydroxamate (MeAHA). Calculations were performed under simulated basic and acidic conditions. Ligand substitution under basic conditions was found to be thermodynamically feasible with an overall calculated change in solvation free energy, ΔGsol = −97 kJ mol−1 using the B3LYP/DGDZVP methodology and a water continuum solvation model. In contrast, an acid-mediated mechanism of ligand substitution was found to be thermodynamically non-feasible. Molecular orbital analysis provides a rationale for the difference in thermodynamic stability between [Zr(C2O4)4]4− and Zr(MeAHA)4. Overall, the DFT calculations are consistent with observed experimental 89Zr-radiolabelling reactions and suggest that computational methods may prove useful in designing novel chelates for increasing the thermodynamic and kinetic stability of 89Zr-complexes in vivo.
 Please wait while we load your content...
Please wait while we load your content...

