
Hydration properties determining the reactivity of nitrite in aqueous solution
Abstract
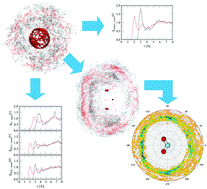
The knowledge of the hydration properties of the nitrite ion is key to understanding its reaction mechanism controlled by solvent effects. Here, ab initio quantum mechanical charge field molecular dynamics was performed to obtain the structural and dynamical properties of the hydration shell in an aqueous solution of nitrite ions, elucidated by data analysis using a molecular approach and an extended quantitative analysis of all superimposed trajectories with three-dimensional alignment (density map). The pattern of the power spectra corresponded to the experimental data, indicating the suitability of the Hartree–Fock method coupled with double-ζ plus polarization and diffuse functional basis sets to study this system. The density maps revealed the structure of the hydration shell, that presented a higher density in the N–O bond direction than in the axis vertical to the molecular plane, whereas the atomic and molecular radial distribution functions provided vague information. The number of actual contacts indicated 4.6 water molecules interacting with a nitrite ion, and 1.5 extra water molecules located in the molecular hydration shell, forming a H-bonding network with the bulk water. The mean residence times for the water ligands designated the strength of the hydration spheres for the oxygen sites, whilst the results for the nitrogen sites over-estimated the number of water molecules from other sites and indicated a weak structure. These results show the influence of the water molecules surrounding the nitrite ion creating an anisotropic hydration shell, suggesting that the reactive sites are situated above and below the molecular plane with a lower water density.
 Please wait while we load your content...
Please wait while we load your content...

