
Large-scale virtual high-throughput screening for the identification of new battery electrolyte solvents: evaluation of electronic structure theory methods†
Abstract
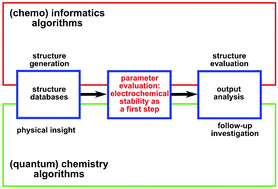
The performance of semi-empirical quantum mechanical (SQM), density functional theory (DFT) and wave function theory (WFT) methods is evaluated for the purpose of screening a large number of molecular structures with respect to their electrochemical stability to identify new battery electrolyte solvents. Starting from 100 000 database entries and based on more than 46 000 DFT calculations, 83 candidate molecules are identified and then used for benchmarking lower-level computational models (SQM, DFT) with respect to higher-level WFT reference data. A combination of SQM and WFT methods is suggested as a screening strategy at the electronic structure theory level. Using a subset of over 11 000 typical organic molecules and based on over 22 000 high-level WFT calculations, several simple models are tested for the prediction of ionization potentials (IPs) and electron affinities (EAs). Reference data are made available for the development of more sophisticated QSPR models.
 Please wait while we load your content...
Please wait while we load your content...

