
Computational modeling of single- versus double-anchoring modes in di-branched organic sensitizers on TiO2 surfaces: structural and electronic properties†
Abstract
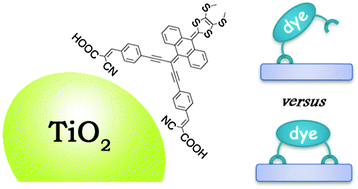
We present a first-principles DFT investigation of the adsorption geometry on the anatase (101) surface of a prototypical di-branched organic dye based on the extended tetrathiafulvalene moiety, incorporating two anchoring cyanoacrylic acid units. Reduced model systems with one and two anchoring groups have been initially studied to investigate the vibrational frequencies related to TiO2 dye adsorption. Our calculations confirm that the reduced systems can be used as reliable models to study the anchoring modes and that the conclusions extracted from the reduced systems can be extrapolated to the entire molecule. A series of molecular structures have been investigated to simulate the anchoring environment in monodentate- and bidentate-like adsorption modes. The comparison between the theoretical results and the available experimental data suggests a di-anchored monodentate adsorption mode as the most probable adsorption structure. Geometry optimizations of the di-branched model system adsorbed on a periodic slab of anatase (101) allowed us to compare the relative stability of different adsorption conformations and led to a di-anchored monodentate mode as the most stable adsorption structure. Furthermore, ab initio molecular dynamics simulations confirmed this structure as the preferred one, providing additional stabilization by effective hydrogen-bonding to surface oxygens and structure distortion from planarity. The analysis of the partial density of states for the prototypical models confirms that the doubly anchored adsorption provides improved electronic properties compared to the singly anchored structures for dye-sensitized solar cell purposes.
 Please wait while we load your content...
Please wait while we load your content...

