
Anisotropy of the n-type charge transport and thermal effects in crystals of a fluoro-alkylated naphthalene diimide: a computational investigation†
 *ab
*ab
Abstract
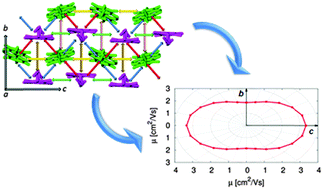
The anisotropy of the n-type charge transport of a fluoro-alkylated naphthalene diimide is investigated in the framework of the non-adiabatic hopping mechanism. Charge transfer rate constants are computed within the Marcus–Levich–Jortner formalism including a single effective mode treated quantum-mechanically and are injected in a kinetic Monte Carlo scheme to propagate the charge carrier in the crystal. Charge mobilities are computed at room temperature with and without the influence of an electric field and are shown to compare very well with previous measurements in single-crystal devices which offer a superior substrate for testing molecular models of charge transport. Thermally induced dynamical effects are investigated by means of an integrated computational approach including molecular dynamics simulations coupled to quantum-chemical evaluation of electronic couplings. It is shown that charge transport occurs mainly in the b,c crystallographic plane with a major component along the c axis which implies an anisotropy factor in very good agreement with the observed value.
 Please wait while we load your content...
Please wait while we load your content...

