
Theoretical study of temperature dependence and Rayleigh scattering properties of chloride hydration clusters
Abstract
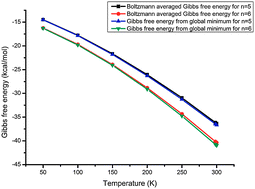
Cl−(H2O)n (n = 5–6) clusters were investigated using a basin hopping (BH) method coupled with density functional theory (DFT). Structures, energetics, thermodynamics, and vibrational frequencies were obtained using high level ab initio calculations. DF-LMP2 (second-order Møller–Plesset perturbation theory using local and density fitting approximations) with an appropriate basis set were employed for final optimization and frequency calculation, which has been benchmarked in a recent study. The global minimum of Cl−(H2O)5 was verified and the new competitive local minimum of Cl−(H2O)6 was offered. Considering the increasing complexity of the large system and the high flexibility of the hydrogen bonding environment, Boltzmann averaged Gibbs free energy was provided taking into account the contributions of local minima on the potential energy surface. Finally, the temperature dependence of the conformational population for isomers of Cl−(H2O)n (n = 5–6) and Rayleigh scattering properties of Cl−(H2O)n (n = 1–6) have been investigated systematically for the first time.
 Please wait while we load your content...
Please wait while we load your content...

