
Accurate predictions of C–SO2R bond dissociation enthalpies using density functional theory methods†
Abstract
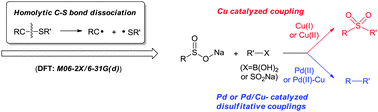
The dissociation of the C–SO2R bond is frequently involved in organic and bio-organic reactions, and the C–SO2R bond dissociation enthalpies (BDEs) are potentially important for understanding the related mechanisms. The primary goal of the present study is to provide a reliable calculation method to predict the different C–SO2R bond dissociation enthalpies (BDEs). Comparing the accuracies of 13 different density functional theory (DFT) methods (such as B3LYP, TPSS, and M05 etc.), and different basis sets (such as 6-31G(d) and 6-311++G(2df,2p)), we found that M06-2X/6-31G(d) gives the best performance in reproducing the various C–S BDEs (and especially the C–SO2R BDEs). As an example for understanding the mechanisms with the aid of C–SO2R BDEs, some primary mechanistic studies were carried out on the chemoselective coupling (in the presence of a Cu-catalyst) or desulfinative coupling reactions (in the presence of a Pd-catalyst) between sulfinic acid salts and boryl/sulfinic acid salts.
 Please wait while we load your content...
Please wait while we load your content...

