
The surface chemistry of NOx on mackinawite (FeS) surfaces: a DFT-D2 study
Abstract
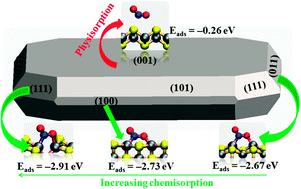
We present density functional theory calculations with a correction for the long-range interactions (DFT-D2) of the bulk and surfaces of mackinawite (FeS), and subsequent adsorption and dissociation of NOx gases (nitrogen monoxide (NO) and nitrogen dioxide (NO2)). Our results show that these environmentally important molecules interact very weakly with the energetically most stable (001) surface, but adsorb relatively strongly onto the FeS(011), (100) and (111) surfaces, preferentially at Fe sites via charge donation from these surface species. The NOx species exhibit a variety of adsorption geometries, with the most favourable for NO being the monodentate Fe–NO configuration, whereas NO2 is calculated to form a bidentate Fe–NOO–Fe configuration. From our calculated thermochemical energy and activation energy barriers for the direct dissociation of NO and NO2 on the FeS surfaces, we show that NO prefers molecular adsorption, while dissociative adsorption, i.e. NO2 (ads) → [NO(ads) + O(ads)] is preferred over molecular adsorption for NO2 onto the mackinawite surfaces. However, the calculated high activation barriers for the further dissociation of the second N–O bond to produce either [N(ads) and 2O(ads)] or [N(ads) and O2(ads)] suggest that complete dissociation of NO2 is unlikely to occur on the mackinawite surfaces.
 Please wait while we load your content...
Please wait while we load your content...

