
Reply to the ‘Comment on “Density functional theory analysis of structural and electronic properties of orthorhombic perovskite CH3NH3PbI3”’ by J. Even et al., Phys. Chem. Chem. Phys., 2014, 10.1039/C3CP55006K†
Abstract
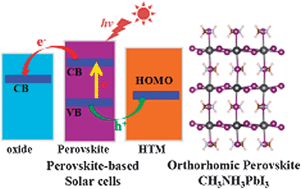
Perovskite CH3NH3PbI3 materials were theoretically investigated using density functional theory (DFT) since they are an important component in novel perovskite-based solar cells. One of the challenges is to accurately describe their electronic structures. As stated in our original paper, the accidental agreement of band gap energies between the traditional DFT calculations and the experimental measurement is “fortuitous”. The disadvantage of traditional DFT can be partially solved by the recent progress made by Even et al. with the consideration of spin–orbit coupling and many-body self-energy corrections. However, the C–N bonding mechanisms in CH3NH3+ cations cannot be deduced from the Bader charge analysis.
 Please wait while we load your content...
Please wait while we load your content...

