
Beyond the molecular orbital conception of electronically excited states through the quantum theory of atoms in molecules†
Abstract
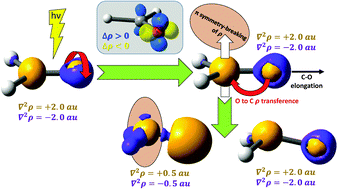
We show that the use of the quantum theory of atoms in molecules (QTAIM) in electronically excited states allows expanding the knowledge that the molecular orbital (MO) framework provides about electronic rearrangements. Despite that historical prejudice seemed to preclude the use of QTAIM beyond the electronic ground state, this paper evidences that QTAIM is versatile enough to deal with excited states. As an example, the paradigmatic n → π* electronic transition of formaldehyde is analyzed. Using QTAIM, an energy partition of excited state energies into atomic and diatomic energies is carried out for the first time. This partition shows that upon electronic excitation the atoms of the CO bond experience a stabilization in their net energies, accompanied by a destabilization in their interaction, a fact which is in accordance with the idea of populating an antibonding π* MO. The associated C–O bond elongation in the nπ* state does not involve a change in the π atomic populations – as one would expect from a π* orbital – but in the σ ones. Moreover, it is also found that the nπ* state is characterized by a weaker C–O interaction energy in comparison to that in the electronic ground state. In order to strengthen this interaction, the electron–electron repulsion between C and O is reduced via a symmetry-breaking of the electron density, causing the C pyramidalization. A topological analysis based on the Laplacian of the electron density and on the electron localization function (ELF) reveals that the n → π* transition can be visualized as a rotation of 90° of the oxygen lone pairs.
 Please wait while we load your content...
Please wait while we load your content...

