
A nine-dimensional global potential energy surface for NH4(X2A1) and kinetics studies on the H + NH3 ↔ H2 + NH2 reaction
Abstract
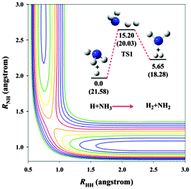
Extensive ab initio calculations of the stationary points in the NH4(X2A1) system are reported using both coupled cluster and multi-reference configuration interaction methods. In addition, more than 100 000 points are generated over a large configuration space and energy range (6 eV) using the explicitly correlated unrestricted coupled cluster method with single, double, and perturbative triple excitations with the augmented correlation-consistent polarized triple zeta basis set (UCCSD(T)-F12a/aug-cc-pVTZ). Using the recently proposed permutation-invariant polynomial neural network (PIP-NN) method, these points are accurately fit to an analytical form with a total root mean squared error (RMSE) of 3.4 meV (0.08 kcal mol−1). Both the abstraction and exchange channels as well as the metastable ammonium radical (NH4) are included in this potential energy surface. Transition-state theory and quasi-classical trajectory calculations have been performed to obtain the rate constants for the abstraction reaction and its reverse. Comparison with available experimental results is satisfactory, providing supporting evidence for the accuracy of the potential.
 Please wait while we load your content...
Please wait while we load your content...

