
Theoretical investigation of the borazine–melamine polymer as a novel candidate for hydrogen storage applications†
Abstract
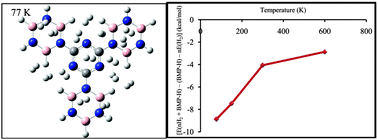
Ab initio calculations and molecular dynamic simulation were employed to study the interaction of molecular hydrogen with the borazine–melamine polymer (BMP) in order to explore its potential for hydrogen storage applications. The calculations were performed using the long range corrected version of density functional theory, the Coulomb-attenuating method (CAM-B3LYP) and the second order Møller–Plesset perturbation theory (MP2). The results showed that the average adsorption energy per hydrogen is about −0.7 and −0.3 kcal mol−1 at the MP2/6-311+G(d,p) and CAMB3LYP/6-311+G(d,p) levels of theory, respectively. The adsorption energies were corrected for the basis set superposition error (BSSE) by the counterpoise method. It was found that the hydrogen storage capacity of the BMP is about 6.49 wt%, which is close to the values reported for the other selected materials for the hydrogen storage in the literature. The maximum number of hydrogen molecules, which were adsorbed by the BMP building block, is about ten. Molecular dynamic simulation was performed to assess the potential of BMP for hydrogen storage.
 Please wait while we load your content...
Please wait while we load your content...

