
Large polarization but small electron transfer for water around Al3+ in a highly hydrated crystal†
Abstract
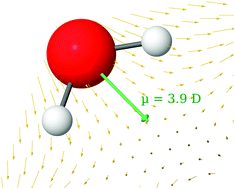
Precise molecular-level information on the water molecule is precious, since it affects our interpretation of the role of water in a range of important applications of aqueous media. Here we propose that electronic structure calculations for highly hydrated crystals yield such information. Properties of nine structurally different water molecules (19 independent O⋯O hydrogen bonds) in the Al(NO3)3·9H2O crystal have been calculated from DFT calculations. We combine the advantage of studying different water environments using one and the same compound and method (instead of comparing a set of independent experiments, each with its own set of errors) with the advantage of knowing the exact atomic positions, and the advantage of calculating properties that are difficult to extract from experiment. We find very large Wannier dipole moments for H2O molecules surrounding the cations: 4.0–4.3 D (compared to our calculated value of 1.83 D in the gas phase). These are induced by the ions and the H-bonds, while other water interactions and the relaxation of the internal water geometry in fact decrease the dipole moments. We find a good correlation between the water dipole moment and the O⋯O distances, and an even better (non-linear) correlation with the average electric field over the molecule. Literature simulation data for ionic aqueous solutions fit quite well with our crystalline ‘dipole moment vs. O⋯O distance’ curve. The progression of the water and cation charges from ‘small clusters ⇒ large clusters ⇒ the crystal’ helps explain why the net charges on all the water molecules are so small in the crystal.
 Please wait while we load your content...
Please wait while we load your content...

