
Aqueous solvation of HgClOH. Stepwise DFT solvation and Born–Oppenheimer molecular dynamics studies of the HgClOH–(H2O)24 complex†
Abstract
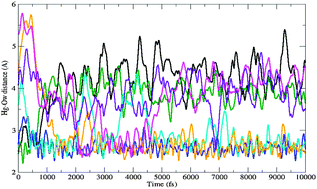
We address the aqueous solvation of HgClOH through a systematic study of stepwise hydration considering the HgClOH–(H2O)n structures with n = 1–24. After calibration of the DFT method, the electronic calculations have been carried out using the B3PW91 exchange–correlation functional. For n < 5 the main geometrical parameters and incremental binding energies are in agreement with counterpoise-corrected MP2/AVTZ static values and BO-MP2 dynamic averages. For n > 15 three direct water–Hg interactions appear during the hydration process and a pentacoordinated trigonal bipyramid apical pattern around Hg is found. 22 water molecules are needed to build the first solvation shell. Unlike microsolvated HgCl2, no stable equatorial trigonal bipyramid was found. Optimizations with the Polarizable Continuum Model lead to structures with extremely large Hg–O(water) distances because of a dominant solvation effect on the explicit water molecules; however, this overestimation diminishes for large values of n. A DFT Born–Oppenheimer molecular dynamics simulation at T = 700 K revealed the stability of the HgClOH–(H2O)24 complex with an average trigonal bipyramid Hg-coordination pattern, in accordance with the static cluster description. After thermalization is achieved, the exchange rate of the Hg-coordinated water molecules is estimated to be ca. 1011 s−1.
 Please wait while we load your content...
Please wait while we load your content...

