
Radical O–O coupling reaction in diferrate-mediated water oxidation studied using multireference wave function theory†
Abstract
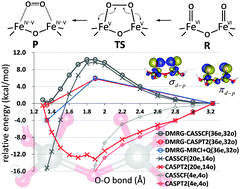
The O–O (oxygen–oxygen) bond formation is widely recognized as a key step of the catalytic reaction of dioxygen evolution from water. Recently, the water oxidation catalyzed by potassium ferrate (K2FeO4) was investigated on the basis of experimental kinetic isotope effect analysis assisted by density functional calculations, revealing the intramolecular oxo-coupling mechanism within a di-iron(VI) intermediate, or diferrate [Sarma et al., J. Am. Chem. Soc., 2012, 134, 15371]. Here, we report a detailed examination of this diferrate-mediated O–O bond formation using scalable multireference electronic structure theory. High-dimensional correlated many-electron wave functions beyond the one-electron picture were computed using the ab initio density matrix renormalization group (DMRG) method along the O–O bond formation pathway. The necessity of using large active space arises from the description of complex electronic interactions and varying redox states both associated with two-center antiferromagnetic multivalent iron–oxo coupling. Dynamic correlation effects on top of the active space DMRG wave functions were additively accounted for by complete active space second-order perturbation (CASPT2) and multireference configuration interaction (MRCI) based methods, which were recently introduced by our group. These multireference methods were capable of handling the double shell effects in the extended active space treatment. The calculations with an active space of 36 electrons in 32 orbitals, which is far over conventional limitation, provide a quantitatively reliable prediction of potential energy profiles and confirmed the viability of the direct oxo coupling. The bonding nature of Fe–O and dual bonding character of O–O are discussed using natural orbitals.
- This article is part of the themed collection: Photosynthesis: From Natural to Artificial
 Please wait while we load your content...
Please wait while we load your content...

