
Self-aggregation mechanisms of N-alkyl derivatives of urea and thiourea†
Abstract
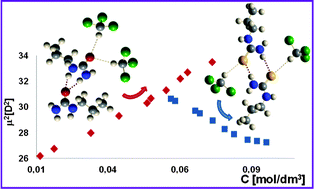
The mechanisms of self-aggregation of N-alkyl and N,N′-dialkyl derivatives of urea and thiourea in weakly polar solvents (chloroform and 1,2-dichloroethane) were examined. The C–H⋯O or C–H⋯S hydrogen bonds formed with these two acidic solvents compete with the N–H⋯O or N–H⋯S hydrogen bonds formed between solute molecules, influencing the self-aggregation of urea derivatives in a particular solvent. The peculiarities of the solvent interactions were discussed and the stronger interaction of chloroform was noted. Aggregation of the N-alkyl derivatives was followed using IR spectroscopy, with two gradual aggregation constants (K1 and K2) determined. The average molecular weight and dipole moments were shown to depend on the concentration, and the form of aggregation was analyzed through the study of the dipole moments. All of the urea derivatives demonstrated an increase in dipole moment with increased concentration, resulting in stronger NH2⋯O hydrogen bond interactions and leading to linear-type aggregation. Contrastingly, the dipole moments of the mono-N-alkyl-substituted thioureas decreased with concentration. Density-functional theory calculation of these processes showed that reliable results could only be obtained if solvent interactions were considered, with a specific combination of local and bulk effects. It was also shown that going from N,N′-disubstituted to N-monoalkyl derivatives the ability to aggregate increases, which is related to a diminished steric hindrance to hydrogen bonding. Finally, it was demonstrated that the mechanisms of self-aggregation depend on the acid–base properties of the solute, hydrogen bonding to the solvent molecules, and steric interactions of the aliphatic chains.
 Please wait while we load your content...
Please wait while we load your content...

