
Theoretical study on the mechanism of Pd(OAc)2 catalyzed dehydrogenative cross-coupling of two heteroarenes†
Abstract
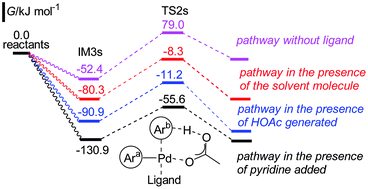
The dehydrogenative cross-coupling between caffeine and 2-formylfuran has been theoretically studied using a DFT method. It is found that the coupling process consists of three steps: 2 consecutive C–H bond activations, and reductive elimination. The mechanism with pyridine added as a ligand is energetically more favored than that with the solvent molecule or the generated acetic acid as or without ligand. Two kinds of pathways involving the prior activation of one of the coupling partners have been performed, and the prior activation of caffeine by the Pd(OAc)2 catalyst, followed by the activation of 2-formylfuran is energetically preferred. The calculated results indicated that the activation of 2-formylfuran is the rate-determining step with an energy barrier of 75.3 kJ mol−1. The interaction of the first activated partner with the catalyst assists the activation of the second partner, and the C–H activation of caffeine is easier than that of 2-formylfuran as the energy barrier is 22.3 kJ mol−1 lower. The above two parameters provide mainly the high selectivity to the cross-coupling product. The mechanism of the dehydrogenative cross-coupling of caffeine and 2-formylthiophen is similar to that described above.
 Please wait while we load your content...
Please wait while we load your content...

