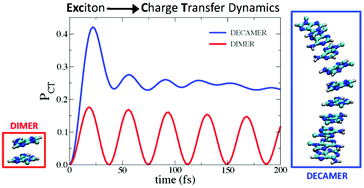
We investigate the quantum dynamics of the internal conversion of excitons into charge transfer (CT) states in single-strand oligomers of adenine (An) of different length (n up to 10 units) excited by a short-time laser pulse. Calculations are based on a model vibronic Hamiltonian whose parameters are fitted to accurate time-dependent density functional theory (TD-DFT) calculations, which was shown to reproduce the experimental absorption spectrum with the increase of n. As a first step, we analyze the impact of the vibrational motion on the population transfer in the dimer, highlighting that it causes loss of coherence and slows down the dynamics. For longer oligomers we resort to a simplified approach considering only electronic states and solving the equation of motion for the density matrix driven by inter-state couplings. In this way we are able also to include phenomenologically dephasing terms that mainly simulate intra-molecular effects, and lifetimes of local excitations mimicking monomer-like decay processes. Relaxation effects, whose role is to drive the system towards the thermal equilibrium allowing population exchange among states, are deliberately not considered here, since the focus is on very short-time dynamics. We consider both the cases of an instantaneous and of a finite-time (full width at half maximum 50 fs) laser pulse. According to our calculations, the photoexcited oligomers exhibit a complex dynamics and CT population rises on a 20–30 fs timescale and it persists even on the picosecond timescale. CT population increases with the length of the oligomer and it is only weakly dependent on the relative stability of CT and exciton states (within a range of 1500 cm−1). The chain length already modifies the photoexcited dynamics for A2 and A4 systems, but this effect saturates for small n so that the A10 oligomer is also representative of longer chains.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
