
Inferring pathway crosstalk networks using gene set co-expression signatures†
Abstract
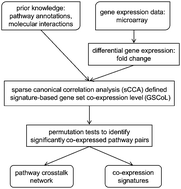
Constructing molecular interaction networks in cells is important for understanding the underlying mechanisms of biological processes. Except for single gene analysis, several gene set-based methods have been proposed to infer pathway crosstalk by analyzing large-scale gene expression data. But most of them take all pathway genes as a whole to infer the crosstalk. Biological evidence suggests that the pathway crosstalk usually occurs between some subsets rather than the whole sets of pathway genes. In this study, we propose a novel method, sGSCA (signature-based gene set co-expression analysis) which can use the co-expression correlations between subsets of pathway genes to infer the pathway crosstalk networks. The method applies sparse canonical correlation analysis (sCCA) to measure the pathway level co-expression and simultaneously obtain the subsets or signature genes that contribute to the co-expression of pathways. On simulated datasets, sGSCA can efficiently detect pathway crosstalk and the corresponding highly correlated signature genes. We applied sGSCA to two cancer gene expression datasets (one for hepatocellular cancer and the other for lung cancer). In the inferred networks, we found several important pathway crosstalks related to the cancers. The identified signature genes also show high enrichment for the cancer related genes. sGSCA can infer pathway crosstalk networks using large-scale gene expression data, and should be a useful tool for systematically studying the molecular mechanisms of complex diseases on both pathway and gene levels at the same time.
 Please wait while we load your content...
Please wait while we load your content...