
An extrapolation method for the efficient calculation of molecular response properties within Born–Oppenheimer molecular dynamics
Abstract
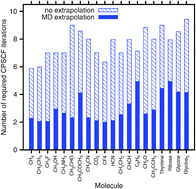
The calculation of molecular response properties in dynamic molecular systems is a major challenge that requires sampling over many steps of, e.g., Born–Oppenheimer molecular dynamics (BO-MD) simulations. We present an extrapolation scheme to accelerate such calculations for multiple steps within BO-MD trajectories or equivalently within other sampling methods of conformational space. The extrapolation scheme is related to the one introduced by Pulay and Fogarasi [Chem. Phys. Lett., 2004, 386, 272] for self-consistent field (SCF) energy calculations. We extend the extrapolation to the quantities within our density matrix-based Laplace-transformed coupled perturbed SCF (DL-CPSCF) method that allows for linear-scaling calculations of response properties for large molecular systems. Here, we focus on the example of calculating
 Please wait while we load your content...
Please wait while we load your content...

