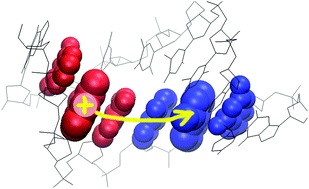
In this work, a fragment-orbital density functional theory-based method is combined with two different non-adiabatic schemes for the propagation of the electronic degrees of freedom. This allows us to perform unbiased simulations of electron transfer processes in complex media, and the computational scheme is applied to the transfer of a hole in solvated DNA. It turns out that the mean-field approach, where the wave function of the hole is driven into a superposition of adiabatic states, leads to over-delocalization of the hole charge. This problem is avoided using a surface hopping scheme, resulting in a smaller rate of hole transfer. The method is highly efficient due to the on-the-fly computation of the coarse-grained DFT Hamiltonian for the nucleobases, which is coupled to the environment using a QM/MM approach. The computational efficiency and partial parallel character of the methodology make it possible to simulate electron transfer in systems of relevant biochemical size on a nanosecond time scale. Since standard non-polarizable force fields are applied in the molecular-mechanics part of the calculation, a simple scaling scheme was introduced into the electrostatic potential in order to simulate the effect of electronic polarization. It is shown that electronic polarization has an important effect on the features of charge transfer. The methodology is applied to two kinds of DNA sequences, illustrating the features of transfer along a flat energy landscape as well as over an energy barrier. The performance and relative merit of the mean-field scheme and the surface hopping for this application are discussed.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


