
Theoretical analysis and quantification of the absorption spectra of uranyl complexes with structurally-related tridentate ligands
Abstract
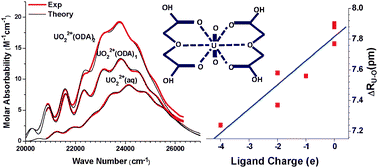
A semi-empirical model of vibronic coupling was developed in the present work to provide a quantitative analysis of partially resolved vibronic structure in uranyl absorption spectra. The studied uranyl complexes share a similar structure but bear different electric charges in the equatorial ligands. Their optical absorption spectra exhibit the common characteristics of charge transfer vibronic transitions of uranyl complexes but fine structures arising from specific vibrational modes are obscured. Accordingly, in application of the Franck–Condon principle of vibronic coupling, our model takes a mode-degenerate approximation. The absorption spectra of uranyl in various ligand environments were calculated and fitted to the experimental data. The semi-empirical approach enabled quantitative evaluation of the electronic energy levels, vibrational frequencies, and vibronic coupling strength. Moreover, the expansion of the U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in the excited states was calculated from the values of the vibronic coupling parameters determined in simulation of the experimental spectra. The calculated results agree very well with the experimentally observed trends in thermodynamic binding constants and structural parameters. A theoretical interpretation is given to the dependence of the UO bond expansion on the charges carried by the uranyl ligand complexes.
O bond in the excited states was calculated from the values of the vibronic coupling parameters determined in simulation of the experimental spectra. The calculated results agree very well with the experimentally observed trends in thermodynamic binding constants and structural parameters. A theoretical interpretation is given to the dependence of the UO bond expansion on the charges carried by the uranyl ligand complexes.
 Please wait while we load your content...
Please wait while we load your content...

