
Computational study of molecular properties with dual basis sets
Abstract
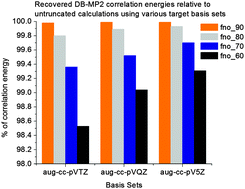
We have studied the performance of dual basis (DB) sets for the evaluation of molecular properties via second order Møller–Plesset perturbation theory (MP2). In addition to savings derived from using a trimmed basis set for the underlying Hartree–Fock (HF) calculation, we pursued a systematic truncation of the virtual subspace for further reductions in computational overhead during the post-HF step. Calculated total energies and molecular properties within the DB framework without virtual space truncation are generally in excellent agreement with full basis calculations. When aug-cc-pV5Z is used as the parent basis, mean absolute error for DB-HF (DB-MP2) total energies of molecules within our test set is 9.7 × 10−5 au (8.0 × 10−5 au) while mean absolute relative errors for static electrical response properties like dipole moments, isotropic dipole polarizabilities and polarizability anisotropies are 0.15% (0.14%), 0.56% (0.72%), and 0.76% (0.83%) respectively. When DB is coupled with virtual space truncation at the MP2 level, the corresponding errors are larger but still within 2% of full basis values.
 Please wait while we load your content...
Please wait while we load your content...

