
Microscopic effects of the bonding configuration of nitrogen-doped graphene on its reactivity toward hydrogen peroxide reduction reaction†
Abstract
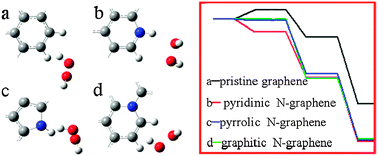
We report a density functional theory (DFT) study of microscopic detailed effects of the bonding configuration of nitrogen-doped graphene (N-graphene) within the carbon lattice (including pyridinic, pyrrolic, and graphitic N) on the reactivity and mechanistic processes of H2O2 reduction reaction. We simulated the adsorption process of H2O2, analyzed the mechanistic processes, and calculated the reversible potential of each reaction step of the H2O2 reduction reaction on N-graphene. The results indicate that the adsorption of H2O2 on the pristine and N-doped graphene surfaces occurs via physisorption without the formation of a chemical bond. When H+ is introduced into the system, a series of reactions can occur, including the breakage of the O–O bond, the formation of an O–C chemical bond between oxygen and graphene, and the creation of water molecules. The results also indicate a decrease in the energy of the system and a positive reversible potential for each reaction step. The calculations of the relative energy of each reaction step and the value of the onset potential for H2O2 reduction reaction suggest that the reactivity of pristine and N-doped graphene has the following order: pyridinic N-graphene > pyrrolic N-graphene > graphitic N-graphene > pristine graphene. We also proposed an explanation based on electrostatic potential calculations for this dependence of the reactivity order on the bond configuration of the doping in N-graphene. The results of this study should help in the atomic-scale understanding of the dependence of the reactivity of N-graphene on its microstructure, inspire the study of various types of heteroatom-doped graphenes to improve their catalytic efficiency, and provide a theoretical framework to analyze their reactivities.
 Please wait while we load your content...
Please wait while we load your content...

