
The insertion of gas molecules into polyhedral oligomeric silsesquioxane (POSS) cages: understanding the energy of insertion using quantum chemical calculations†
Abstract
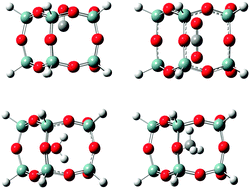
Density functional theory (DFT) calculations using the B3LYP and ωB97XD functionals and Møller–Plesset (MP2) calculations have been used to determine the energy of insertion of seven molecules (CO2, N2, O2, CO, CH4, He and H2O) into two sizes of polyhedral oligomeric silsesquioxane (POSS) cages, T10 and T12. Geometry optimization results of each molecule inserted into the center of the POSS cage and transition-energy searches of the molecules in the largest face of both T10 and T12 (identified as the Si5 face) are discussed. The main factors that affect the energy of a molecule in a particular POSS cage are the size (number of atoms) and shape of the POSS cage and the strength of dispersion and electrostatic interactions. The largest of the molecules, CO2 and CH4, have the largest interaction energies within the center and the face of the T10 and T12 cages. Interaction energies for the diatomic molecules increase in the order H2 < O2 < N2 ∼ CO and differences between interaction energies can be attributed, in part, to differences in electron transfer between the cage and molecule. Electrostatic interactions also play a significant role in determining the interaction energy. For example, H2 and He are only slightly charged when inside the POSS cages, providing little repulsive or attractive interactions, while polar CO provides significant repulsive interaction. In the case of water, the interaction energies are low because of hydrogen bonding between the water hydrogen atoms and the POSS oxygen atoms. The interaction energies of the absorbates at the center of T12 are typically smaller than for T10 due to the larger cavity size of T12. The Si5 face has the same basic structure for both T10 and T12 cages and, consequently, the interaction energies of the absorbate molecules at the face are similar. Interaction energies obtained using MP2 calculations are lower than those obtained from DFT calculations depending on the choice of the functional. For example, use of the ωB97XD functional that treats dispersion interactions more completely than the B3LYP functional, produces values closer to the MP2 values. These results, therefore, suggest that dispersion is important in the interaction between small molecules and POSS cages.
 Please wait while we load your content...
Please wait while we load your content...

