
First-principles study of the formation and migration of native defects in LiNH2BH3†
Abstract
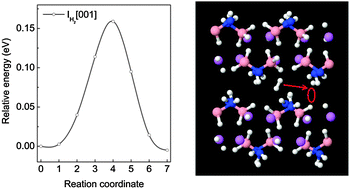
First-principles calculations based on density functional theory were carried out to investigate the formation and migration of native defects in LiNH2BH3. The structural properties and formation energies of H, Li, N and B related defects in various charge states were examined. Our analysis showed that the dominant atomic H defects are positively and negatively charged H interstitial (IH+ and IH−). The Li related defects are dominated by positively charged Li interstitial (ILi+) and negatively charged Li vacancy (VLi−). For N related defects, the energetically favorable defects are positively and negatively charged NH2 interstitial (INH2+ and INH2−), while B related defect is dominated by neutral BH3 interstitial (IBH30). Further results indicated that the neutral H2 interstitial (IH2) has the lowest formation energy (0.31 eV), suggesting that the major defect in LiNH2BH3 is IH2. Investigation of migration processes of the defects showed that the migration barriers for V(B)H+ (positively charged H vacancy on a B–H site), IH+ and IH− are relatively high (0.50–0.68 eV), whereas moderate diffusion barriers are presented for V(N)H− (negatively charged H vacancy on a N–H site) and ILi+ (0.29 and 0.32 eV, respectively). The VLi− and IH2 defects can migrate with low energy barriers of 0.13 and 0.16 eV, respectively. With a low activation energy of 0.47 eV, IH2 is the major diffusive species in LiNH2BH3. Our calculation results further suggest that the creation of the V(N)H−, ILi+ and VLi− defects is the rate-limiting step for their transportation in LiNH2BH3.
 Please wait while we load your content...
Please wait while we load your content...

