
Deactivation mechanism of AuCl3 catalyst in acetylene hydrochlorination reaction: a DFT study
Abstract
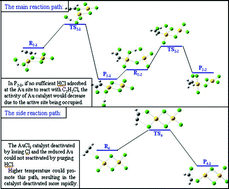
The deactivation mechanism of AuCl3 catalyst in the reaction of acetylene hydrochlorination was studied by using AuCl3 dimer model and the density functional theory (DFT) method. Four possible paths for the acetylene hydrochlorination reaction catalyzed by AuCl3 were illustrated with corresponding transition states. The activation free energies and reaction rate constants of the four paths were also analyzed. It is apparent that when HCl and C2H2 coadsorbed on the AuCl3 dimer, the C2H2 was co-catalyzed by HCl and the AuCl3 dimer to produce C2H3Cl and the reaction energy barrier was as low as 23.35 kcal mol−1. If the HCl in the gas phase could not adsorb on the Au site within the set time, the intermediate chlorovinyl was difficult to desorb from the AuCl3 catalyst as its desorption energy was as high as 41.336 kcal mol−1. As the reaction temperature increased, C2H2 became easier to be adsorbed on the AuCl3 catalyst prior to HCl, which resulted in the side reaction and the rapid deactivation of the AuCl3 dimer due to the loss of Cl atoms. Our calculations are necessary for us to clearly understand the experimental results, which indicate a great dependence of activity and stability of AuCl3 catalysts on the HCl : C2H2 ratio as well as the temperature.
 Please wait while we load your content...
Please wait while we load your content...

