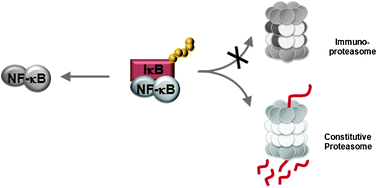
The discovery of NF-κB signaling pathways has greatly enhanced our understanding of inflammatory and immune responses. In the canonical NF-κB pathway, the proteasomal degradation of IκBα, an inhibitory protein of NF-κB, is widely accepted to be a key regulatory step. However, contradictory findings have been reported as to whether the immunoproteasome plays an obligatory role in the degradation of IκBα and activation of the canonical NF-κB pathway. Such results were obtained mainly using traditional gene deletion strategies. Here, we have revisited the involvement of the immunoproteasome in the canonical NF-κB pathway using small molecule inhibitors of the immunoproteasome, namely UK-101 and LKS01 targeting β1i and β5i, respectively. H23 and Panc-1 cancer cells were pretreated with UK-101, LKS01 or epoxomicin (a prototypic inhibitor targeting both the constitutive proteasome and immunoproteasome). We then examined whether these pretreatments lead to any defect in activating the canonical NF-κB pathway following TNFα exposure by monitoring the phosphorylation and degradation of IκBα, nuclear translocation of NF-κB proteins and DNA binding and transcriptional activity of NF-κB. Our results consistently indicated that there is no defect in activating the canonical NF-κB pathway following selective inhibition of the immunoproteasome catalytic subunits β1i, β5i or both using UK-101 and LKS01, in contrast to epoxomicin. In summary, our current results using chemical genetic approaches strongly support that the catalytic activity of the immunoproteasome subunits β1i and β5i is not required for canonical NF-κB activation in lung and pancreatic adenocarcinoma cell line models.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
