
Revisiting the homology modeling of G-protein coupled receptors: β1-adrenoceptor as an example†
Abstract
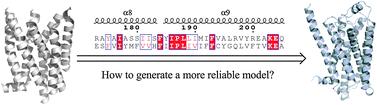
G-protein coupled receptors (GPCRs) are recognized to constitute the largest family of membrane proteins. Due to the disproportion in the quantity of crystal structures and their amino acid sequences, homology modeling contributes a reasonable and feasible approach to GPCR theoretical coordinates. With the brand new crystal structures resolved recently, herein we deliberated how to designate them as templates to carry out homology modeling in four aspects: (1) various sequence alignment methods; (2) protein weight matrix; (3) different sets of multiple templates; (4) active and inactive state of templates. The accuracy of models was evaluated by comparing the similarity of stereo conformation and molecular docking results between models and the experimental structure of Meleagris gallopavo β1-adrenergic receptor (Mg_Adrb1) that we desired to develop as an example. Our results proposed that: (1) Cobalt and MAFFT, two algorithms of sequence alignment, were suitable for single- and multiple-template modeling, respectively; (2) Blosum30 is applicable to align sequences in the case of low sequence identity; (3) multiple-template modeling is not always better than single-template one; (4) the state of template is an influential factor in simulating the GPCR structures as well.
 Please wait while we load your content...
Please wait while we load your content...