
Electronic properties and 4f → 5d transitions in Ce-doped Lu2SiO5: a theoretical investigation
Abstract
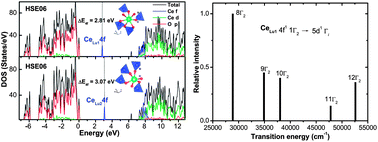
The electronic properties and the 4f → 5d transitions of the dopant Ce3+ ions located at the two crystallographic lutetium sites of Lu2SiO5 (LSO) are investigated using the hybrid density functional theory (DFT) with the HSE06 functional and the wavefunction-based embedded cluster methods, respectively. The HSE06 calculations give a band gap of 6.35 eV for LSO, which agrees well with the reported experimental values between 6.4 and 6.8 eV. It is found that Ce3+ prefers strongly to occupy the seven-coordinated (Lu1) site over the six-coordinated (Lu2) one. The energy gaps between the occupied Ce3+ 4f band and the valence band maximum of the host are predicted to be 2.81 and 3.07 eV for CeLu1 and CeLu2 substitutions, respectively, which are close to the experimental data of 2.6–2.9 eV. Based on the wavefunction-based CASSCF/CASPT2 embedded cluster calculations for the energies of the Ce3+ 4f1 and 5d1 levels, the experimentally observed 4f → 5d transition bands are identified in association with the two substitutions. The predicted transition energy and intensity patterns for CeLu1 substitution are in fairly good agreement with those of the experimental absorption spectrum. The variations of the two lowest 4f → 5d transition energies with the substitutions are finally discussed in terms of the changes of the centroid-energy difference and the crystal-field splitting with the local coordination geometries.
 Please wait while we load your content...
Please wait while we load your content...