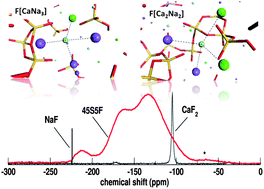
Fluoride-containing bioactive glasses are attracting particular interest in many fields of dentistry and orthopedics because they combine the bone-bonding ability of bioactive glasses with the anticariogenic protection provided by fluoride ions. Since the biomedical applications of these materials critically depend on the release of ionic species in the surrounding physiological environment, a deep knowledge of their environments is required. In this paper, density functional theory calculations and spin effective Hamiltonians have been employed to analyse the NMR signatures of the various environments of 19F, 29Si, 31P and 23Na atoms in fluorinated bioglasses structural models previously generated by Car–Parrinello molecular dynamics simulations. Comparison with experimental spectra expressly recorded in this work shows a good agreement and allows the enlightenment of some longstanding issues about the atomic structure of fluorinated bioglasses, such as the presence of Si–F and Si–O–P bonds. In particular, it is shown that Si–F bonds cannot be resolved by using MAS NMR experiments only, and 29Si{19F} REDOR experiments, that probes directly spatial proximities among atoms, must be employed. Our results show that F is coordinated entirely to the modifier ions Na and Ca, and that no Si–F bonds are present in the real glass structure. Thus, the addition of fluorine to the 45S5 Bioglass® increases the polymerization of the silicate network by removing modifiers from the siliceous matrix and reducing its reactivity. Finally, the computed isotropic chemical shifts of the various environments of phosphorus show that, if present, Si–O–P bonds should be clearly noticeable in the 31P static NMR experimental spectrum. Instead, the latter show that P is present as isolated orthophosphate units and does not enter into the siliceous matrix by forming Si–O–P bonds as conjectured by molecular dynamics simulations.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
