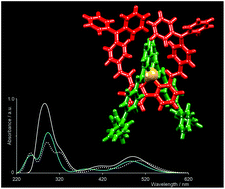
The structures and properties of the homoleptic copper(I) complexes [Cu(1)2][PF6] and [Cu(2)2][PF6] (1 = 6,6′-dimethyl-2,2′-bipyridine, 2 = 6,6′-bis{2-[4-(N,N′-diphenylamino)phenyl]ethenyl}-2,2′-bipyridine) are compared, and a strategy of ligand exchange in solution has been used to prepare eight TiO2 surface-bound heteroleptic complexes incorporating ligands with bpy metal-binding domains and carboxylate or phosphonate anchoring groups. The presence of the extended π-system in 2 significantly improves dye performance, and the most efficient sensitizers are those with phosphonate or phenyl-4-carboxylate anchoring units; a combination of [Cu(2)2]+ with the phosphonate anchoring ligand gives a very promising performance (η = 2.35% compared to 7.29% for standard dye N719 under the same conditions). The high-energy bands in the electronic absorption spectrum of [Cu(2)2]+ which arise from ligand-based transitions dominate the spectrum, whereas that of [Cu(1)2]+ exhibits both MLCT and ligand π* ← π bands. Both [Cu(1)2][PF6] and [Cu(2)2][PF6] are redox active; while the former exhibits both copper-centred and ligand-based processes, [Cu(2)2][PF6] shows only ligand-based reductions. Results of TD-DFT calculations support these experimental data. They predict an electronic absorption spectrum for [Cu(1)2]+ with an MLCT band and high-energy ligand-based transitions, and a spectrum for [Cu(2)2]+ comprising transitions involving mainly contributions from orbitals with ligand 2 character. We have assessed the effects of the atomic orbital basis set on the calculated absorption spectrum of [Cu(1)2]+ and show that a realistic spectrum is obtained by using a 6-311++G** basis set on all atoms, or 6-311++G** on copper and 6-31G* basis set on all other atoms; a smaller basis set on copper leads to unsatisfactory results. Electronic absorption spectra of six heteroleptic complexes have been predicted using TD-DFT calculations, and the transitions making up the dominant bands analysed in terms of the character of the HOMO–LUMO manifold. The calculational data reveal dominant phosphonate ligand character in the LUMO for the dye found to function most efficiently in practice, and also reveal that the orbital character in the HOMOs of the two most efficient dyes is dominated by the non-anchoring ligand 2, suggesting that ligand 2 enhances the performance of the sensitizer by minimizing back-migration of an electron from the semiconductor to the dye.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


