
Ab initio calculations of the melting temperatures of refractory bcc metals
Abstract
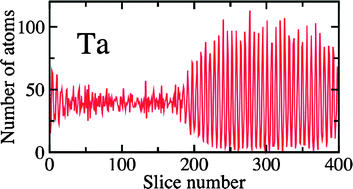
We present ab initio calculations of the melting temperatures for bcc metals Nb, Ta and W. The calculations combine phase coexistence molecular dynamics (MD) simulations using classical embedded-atom method potentials and ab initio density functional theory free energy corrections. The calculated melting temperatures for Nb, Ta and W are, respectively, within 3%, 4%, and 7% of the experimental values. We compare the melting temperatures to those obtained from direct ab initio molecular dynamics simulations and see if they are in excellent agreement with each other. The small remaining discrepancies with experiment are thus likely due to inherent limitations associated with exchange–correlation energy approximations within density-functional theory.
 Please wait while we load your content...
Please wait while we load your content...

